Overview
The Open Catalyst Challenge 2023 invites participants to help address the pressing challenges faced by the world due to energy scarcity and climate change. In this area, a critical problem is the discovery of new catalysts for driving efficient and carbon neutral means for energy storage and conversion. One of the difficulties with finding novel catalysts is that the number of candidates is far too large to test experimentally, so computational screening has begun to play a critical role in the design process. An important quantity in screening catalysts is the adsorption energy, i.e. how strongly a molecule referred to as the “adsorbate” binds to the catalyst’s surface. The rate of the chemical reaction, a value of high practical importance, is then commonly correlated with adsorption energies. To move closer to practical catalyst screening applications, the goal of this year’s challenge is to predict the adsorption energy (global minimum binding energy) given an adsorbate and a catalyst surface (Lan et al., 2022).
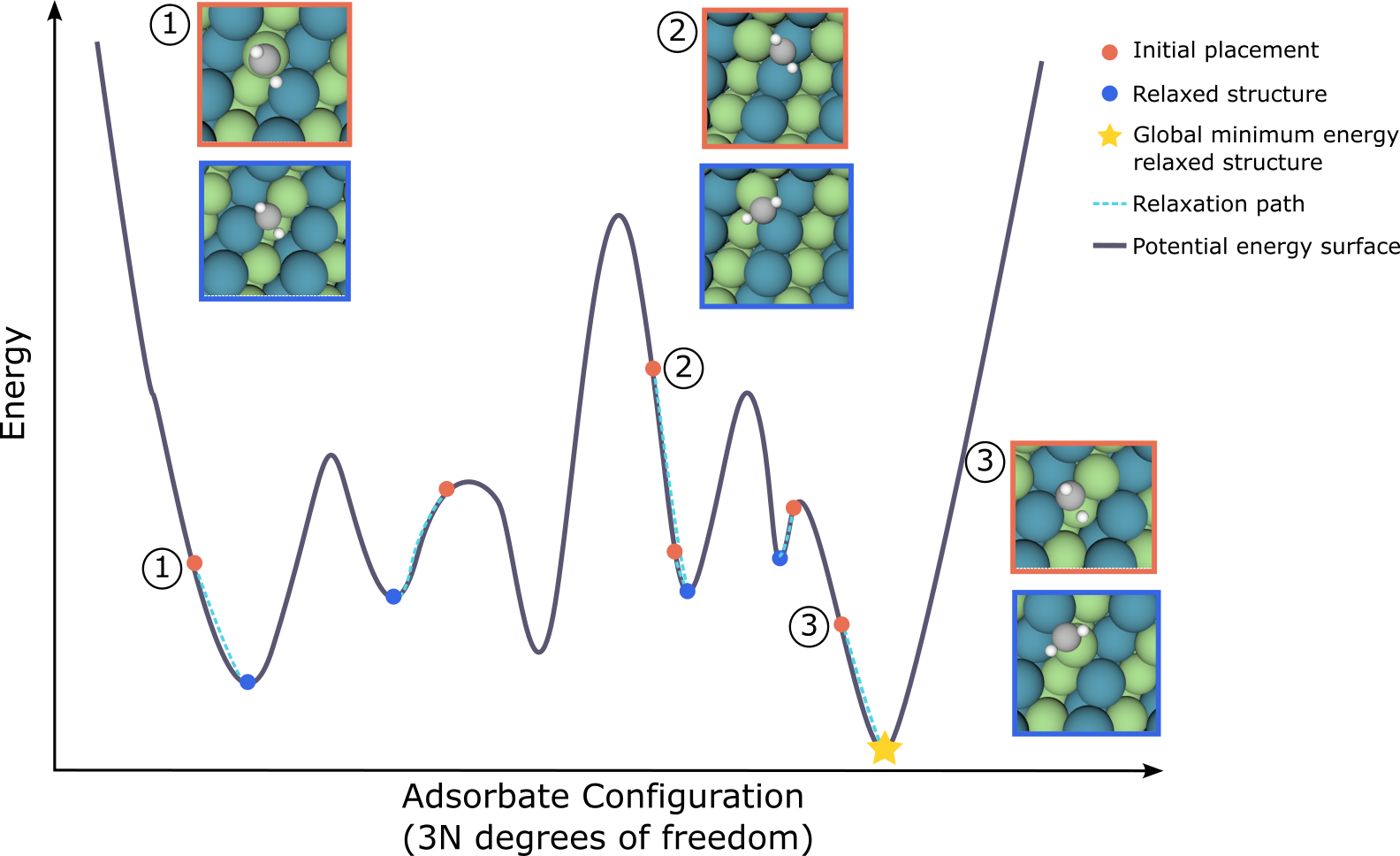
Over the past two years the Open Catalyst Challenge has focused on the central task of relaxed (local minimum) energy prediction. This year’s task of determining the adsorption energy (global minimum) will require relaxed energy prediction as a subtask. In the figure below it is illustrated that different initial adsorbate-surface configurations, unique 3D structures, lead to different local energy minima through relaxation. For this challenge, the energy of the global minimum relaxed structure (noted with a star) is the target, rather than any of the other relaxations which find local minima. Participants need to identify the minimum energy structure and its corresponding energy for a given adsorbate-surface combination. By predicting the global minimum energy (adsorption energy) accurately, the catalyst's impact on the overall rate of a chemical reaction may be estimated; a key factor in filtering potential catalyst materials and addressing the world's energy needs.
This is the 3rd edition of the Open Catalyst Challenge. The 1st and 2nd editions were held at NeurIPS 2021 and 2022, respectively. More details about the previous challenge can be found here.
Dates
Jun 26, 2023: Competition announced
Sep 20, 2023: Challenge test dataset release
Oct 13 Oct 20, 2023: Submission deadline
Oct 30, 2023: Winners notified
Nov 17, 2023: Presenters' slides and videos due
Dec 16, 2023: Results Presentation @ NeurIPS AI for Science Workshop 2023
NeurIPS session
The Open Catalyst Challenge session of the AI for Science Workshop will be held on Saturday, December 16th from 10:05 - 11:10am CT in Hall C2.
Schedule
| 10:05 - 10:25am CT | Challenge introduction, results, and analysis |
Brandon Wood, Muhammed Shuaibi
(Open Catalyst Project) 

|
| 10:25 - 10:35am CT | Winner's talk |
Xinyu Li, Zhen Zhang, Anton van den Hengel, Javen Qinfeng Shi
(The University of Adelaide, Australian Institute of Machine Learning) 



|
| 10:35 - 10:45am CT | Runner-up's talk |
Luigi Bonati, Simone Perego, Pedro Buigues, Pietro Novelli, Riccardo Grazzi, Massimiliano Pontil
(Italian Institute of Technology) 





|
| 10:45 - 10:50am CT | Lightening LLM talk |
Janghoon Ock, Rishikesh Magar, Akshay Antony, Amir Barati Farimani
(Carnegie Mellon University, Deep Apple Therapeutics, Arcbest Technologies) 



|
| 10:50 - 11:10am CT | Discussion | Attendees, participants, organizers |
Challenge results
The Open Catalyst Challenge received 21 submissions in total from 6 teams. All submissions were evaluated on the test-challenge-2023 dataset split consisting of 200 adsorbate-surface combinations.
Team CausalAI won the challenge with a success rate of 46% on the test set.
| Rank | Team | Success Rate (%) |
|---|---|---|
| 1 |
CausalAI Xinyu Li1,2, Zhen Zhang1,2, Anton van den Hengel1,2, Javen Qinfeng Shi1,2 1=The University of Adelaide, 2=Australian Institute of Machine Learning |
46.0 |
| 2 |
The Italian Job Luigi Bonati, Simone Perego, Pedro Buigues, Pietro Novelli, Riccardo Grazzi, Massimiliano Pontil Italian Institute of Technology |
39.5 |
| 3 |
Robo Space |
38.5 |
| 3 |
UCB ASK |
22.5 |
| 5 |
LLM-JAR Janghoon Ock1, Rishikesh Magar2, Akshay Antony3, Amir Barati Farimani1 1=Carnegie Mellon University, 2=Deep Apple Therapeutics, 3=Arcbest Technologies |
5.0 |
| 6 |
AI Eng |
0.50 |
Adsorption Energy Task
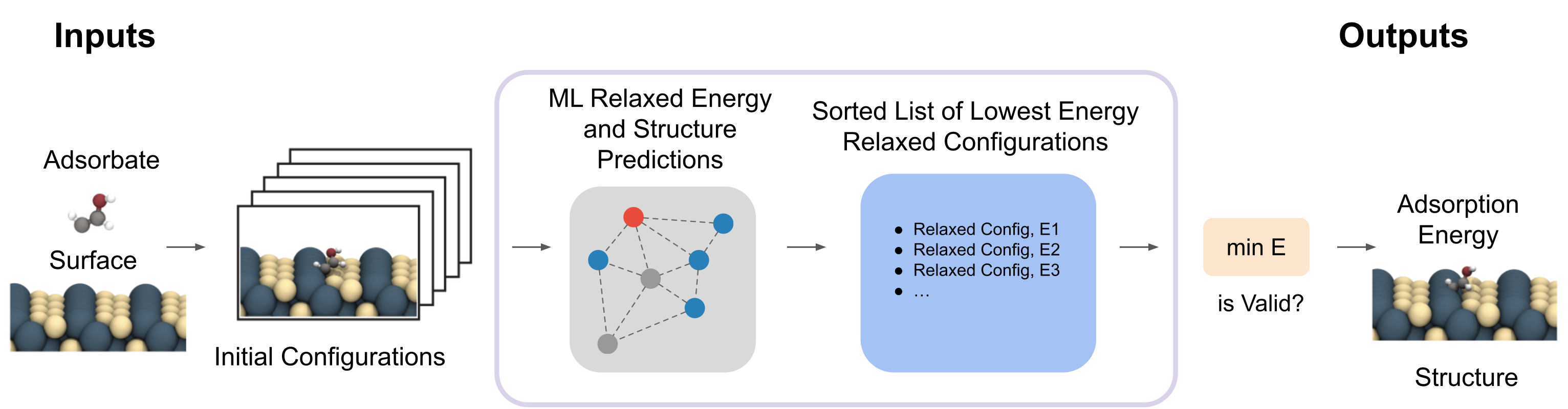
The adsorption energy is traditionally found by first enumerating a large set of initial placements of the adsorbate on the catalyst’s surface. Each of the initial structures are then relaxed using DFT. The adsorption energy is the minimum energy across all of the relaxed structures. This very computationally expensive process typically requires hundreds of DFT calculations to converge (hours or days of compute per relaxation) and forms the basis of most computational catalysis efforts.
In this challenge, participants will be asked to find the adsorption energy for 200 adsorbate/catalyst combinations. For each combination, a set of ~100 initial structures will be released. The goal is to identify the minimum energy relaxed structure and its corresponding energy for each adsorbate/catalyst combination. Additionally, we will release ML relaxed structures (from our top 3 models trained on the OC20-S2EF-2M dataset) for all initial structures provided. Releasing ML relaxed structures allows participants to focus on energy predictions — an area where improvements are needed. As a result the task can be simply restated as, given a set of ML relaxed structures predict their energies and identify the global minimum energy structure and its corresponding energy (adsorption energy). This would be done across all 200 adsorbate/catalyst combinations.
If desired, participants may also choose to perform their own relaxations or use other optimization techniques to increase their chances of finding the global minimum energy and structure.
Additional Areas of Innovation
We described one straightforward approach above, but there could be a host of other ways of solving this year's challenge task. We outline a few below and participants are encouraged to explore these (or any others not listed here)!
S2EF Models
One alternative approach is an end-to-end relaxation method that may provide better relaxed structures than the publicly released relaxed structures provided as part of the challenge. Models developed here would iteratively estimate atomic forces and update atomic positions until a relaxed structure is reached and finally predict the energy of that state. The lowest energy across relaxed structures can then be computed for a given adsorbate-surface combination.
Transfer Learning
Earlier this year, along with the AdsorbML paper, we introduced the OC20-Dense dataset, which includes ~1,000 adsorbate-catalyst surface systems from the OC20 validation set. For each system, the dataset has ~100 configurations of the adsorbate on the catalyst surface. While this dataset was originally released as a way to validate the practical usefulness of OC20 models for predicting the adsorption energy, it provides a rich set of configurations that could be useful for training to predict forces and energies. A portion of this dataset will be made available for training in this year's competition. Fine-tuning pretrained OC20 checkpoints on the new OC20-Dense datasets could provide more accurate models.
Initial Structure Generation
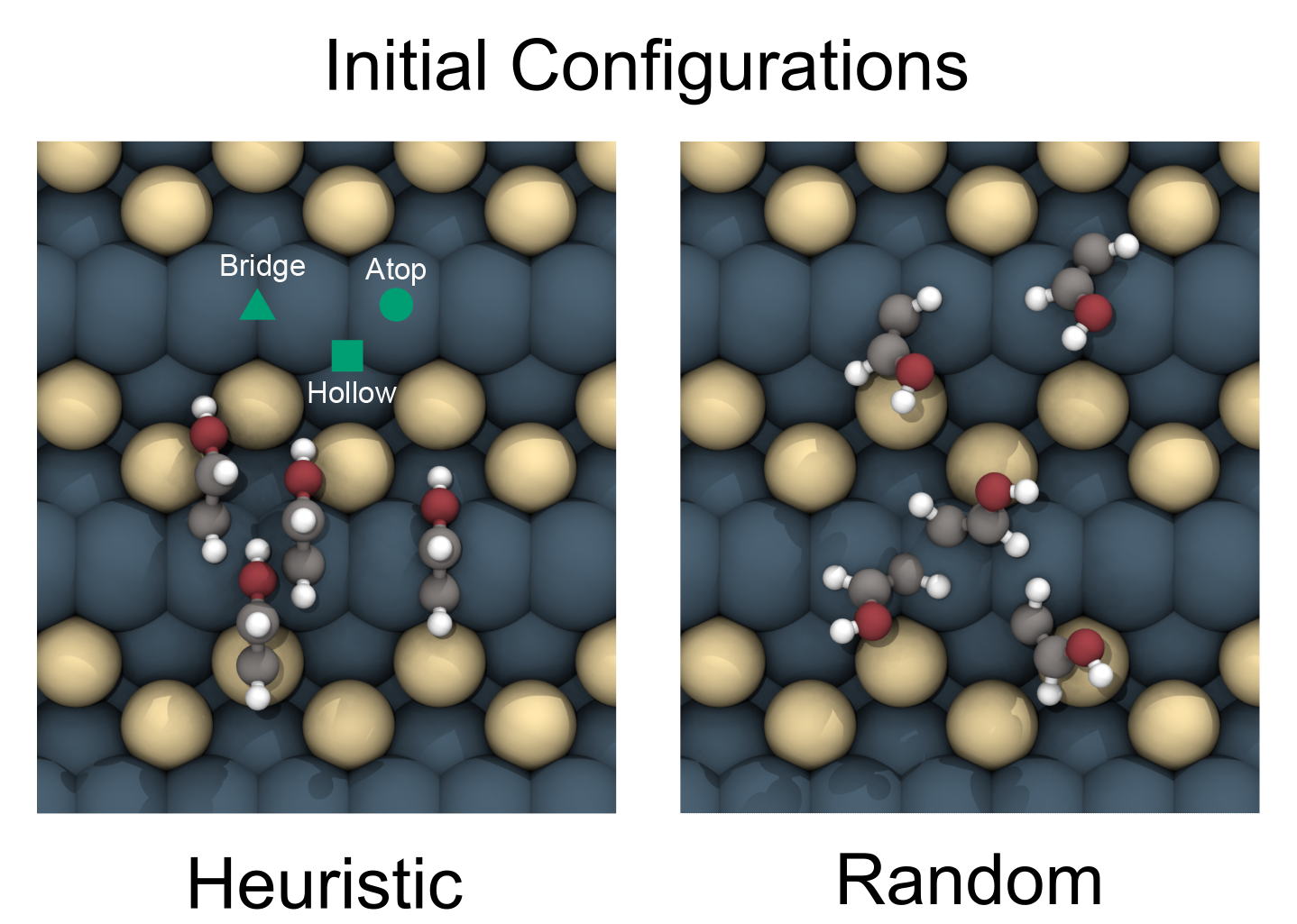
For each of the unique adsorbate-catalyst surfaces, we will release ~100 initial adsorbate structures of that system. Typically, local minima exist when the adsorbate sits on top of a surface atom (atop), between two surface atoms (bridge), or at the center of three or more surface atoms (hollow). We refer to these as "heuristic" placements of the adsorbate on the catalyst surface. For clarity, heuristic placements are highlighted in green in the figure below: atop - circle, bridge – triangle, and hollow – square. To add to the configuration space, we uniformly sample sites on the surface at random with the adsorbate placed on each of those sites with a random rotation along the z-axis. There may be additional opportunity to find even more favorable configurations. Participants are encouraged to generate additional initial placements using any method they like. An overview of how we generated our initial structures can be found as part of the AdsorbML notebook tutorial.
Global Optimization Strategies
Although we supplied initial structures that support dense enumeration as an approach to determine the global minimum, there is opportunity to adopt alternative methods which more directly or efficiently seek to determine the global minimum. This is an important topic in related fields such as protein folding and crystal structure /cluster prediction. Participants are free to explore global optimization strategies.
Large Language Models
The use of pre-trained Large Language Models (LLMs) such as Llama 2 will be allowed. Any additional training or fine-tuning should only use the permitted data listed below in the dataset section. Additionally, these pre-trained LLMs may be classified separately from the ML potentials trained from scratch when determining winners.
Evaluation
All submissions to the Open Catalyst Challenge will be made to the EvalAI server. We will run DFT single-points to evaluate submissions on the following metric:
Success Rate (SR): the percentage of valid predicted adsorption energies within 0.1 eV, or lower, of the DFT-computed ground truth adsorption energy.
A prediction is considered valid under the following conditions:
- The predicted energy is within 0.1 eV of the DFT evaluated energy of the predicted structure.
- Predicted structures do not violate the following physical constraints:
- Desorption: The adsorbate molecule not binding to the surface.
- Dissociation: The adsorbate molecule breaking into different atoms or molecules.
- Surface Mismatch: Surfaces have changed significantly from the corresponding relaxed clean surface.
A more detailed discussion on physical constraints can be found in our AdsorbML paper. Additionally, code is provided to help process predicted ML structures correctly and ensure validity. The image below illustrates the different constraints that need to be avoided.

One unique difference in this year’s challenge is the use of DFT to validate predicted structure and energy predictions. We do not expect participants to run DFT calculations as part of their development process. Models that do well on the S2EF Energy MAE metric are likely to satisfy the energy validation criteria upon submission.
The challenge will have a single track, wherein participants are allowed to train on the OC20-IS2RE/S dataset (size 460k), the OC20-S2EF 2M dataset, and/or OC20-Dense-ID (20k IS2RE, 3M S2EF) dataset. We acknowledge that resource availability may become a bottleneck for some participants and hence will not be accepting entries that train on splits other than the ones described above. The use of pre-trained checkpoints are permitted but shall only come from models trained on datasets detailed in the above table.
The table below summarizes accuracies for relaxation-based baseline approaches. Results are evaluated against the OC20-Dense OOD subsplit made available for validation. As ML relaxed structures will be made available for the provided 2M models, these results provide a baseline for performance. Models trained on S2EF-All+MD (in red) improves accuracies further but due to prohibitively expensive training costs are not allowed for challenge entrants.
| Model | Approach | Training dataset | Success Rate |
|---|---|---|---|
|
GemNet-OC (paper, code) |
Relaxation | S2EF-2M | 30.5% |
|
SCN (paper, code) |
Relaxation | S2EF-2M | 46.5% |
|
eSCN (paper, code) |
Relaxation | S2EF-2M | 40.5% |
|
GemNet-OC-L (paper, code) |
Relaxation | S2EF-All+MD | 37.5% |
|
SCN-L (paper, code) |
Relaxation | S2EF-All+MD | 52.0% |
|
eSCN-L (paper, code) |
Relaxation | S2EF-All+MD | 50.5% |
Dataset
Summary of permitted training data
| Name | Description | Length | Download |
|---|---|---|---|
| OC20-S2EF-2M | OC20 structure to energy and forces (S2EF) data for the 2M split. Does not include dense sampling i.e. there is only one adsorbate placement per surface. | 2,000,000 | Follow S2EF instructions here to download/preprocess the 2M split |
| OC20-Dense-S2EF-ID | OC20-Dense S2EF data for the In-Domain split. Densely sampled S2EF data across 244 adsorbate-surface combinations. | 2,997,635 | LMDB file: oc20dense_s2ef_train.tar.gz |
| OC20-IS2RE/S | OC20 initial structures to relaxed energies/structures (IS2RE/S) data. Does not include dense sampling. | 460,328 | Follow IS2R* instructions here to download |
| OC20-Dense-IS2RE-ID | OC20-Dense IS2RE data for the In-Domain split. Densely sampled IS2RE data across 244 adsorbate-surface combinations. | 15,450 | LMDB file: oc20dense_is2re_train_v2.tar.gz |
OC20 contains approximately ~1.2M DFT relaxations. Due to its significant scale, the dataset required over ~200M hours of compute to generate. Computation was performed on servers Meta has committed to be 100% supported by renewable energy since 2020. Each relaxation contains a series of structures as the atoms move from an initial structure to a relaxed structure obtained through a standard local minimizer built in the computational chemistry code. Structures contain the atoms corresponding to the adsorbate and catalyst. The initial structures are heuristically determined and the relaxed structures correspond to a state in which the atoms are at a local energy minima. Since each step in the relaxation may be used for training / evaluation, the total number of simulation points is over ~264M! The largest training split has ~134M simulation points. However, for this challenge, only the 2M dataset may be used for training. For each structure, DFT computed system energies, per-atom forces and per-atom positions are available as annotations.
As part of our recent AdsorbML work we also introduced the OC20-Dense dataset. OC20-Dense is constructed to closely approximate the adsorption energy for a particular adsorbate-surface combination. To accomplish this, a dense sampling of initial adsorption configurations is necessary. OC20-Dense consists of ∼1,000 unique adsorbate-surface combinations from the OC20 validation set. A uniform sample is taken from each of the validation splits (In-Domain (ID), Out-of-Domain (OOD) Adsorbate, OOD Catalyst, OOD Both) to explore the generalizability of models on this task. For each adsorbate-surface combination, ~100 configurations were sampled. While this dataset was originally released as a way to validate the practical usefulness of OC20 models for adsorption energy calculations, it provides a rich set of configurations that could be useful for training. The In-Domain portion of OC20-Dense will be made available for training in this year’s competition, with a 200 system subset of the OOD splits available for validation. The table below provides download links for the portion of OC20-Dense data permitted for the competition.
Validation data
We provide OC20-Dense validation data to test the performance of your approach for determining the global minimum energy and structure for a particular adsorbate-surface combination. This includes densely sampled initial structures for 200 adsorbate-surface combinations (~ 15k structures in total) in LMDB format to make predictions on and the DFT target file for the evaluation of success rate. Additionally, we provide downloads to DFT outputs as ASE trajectories, including clean surface (surface without any adsorbate) trajectories to be used for filtering physical constraints and a dictionary mapping of LMDB ids to metadata information.
| Name | Description | Download |
|---|---|---|
| OC20-Dense-OOD-val | 15,256 initial structures corresponding to 200 random systems of the OC20-Dense Out-of-Domain (OOD) splits to be used for validation. | LMDB file: oc20dense_ood_val.tar.gz |
| OC20-Dense-OOD-val-ase-format | ASE-compatible atoms object format of "OC20-Dense-OOD-val" (see above). | oc20dense_val_ase.tar.gz |
| OC20-Dense-OOD-val-trajectories | ASE trajectories for converged DFT outputs for the 200 random systems to be used for validation. Note - all configurations may not be available due to removing anomalies or failed DFT calculations. Clean surfaces are also provided for anomaly detection. | oc20dense_val_trajectories.tar.gz |
| OC20-Dense-OOD-val-targets | DFT targets to be used for evaluating the validation subsplit. File should be used for the command line argument --dft-targets here. | oc20dense_val_targets.pkl |
| OC20-Dense-mapping | Dictionary containing a mapping of LMDB sid to general metadata information. Unique system identifiers for an adsorbate/surface combination can be used to map the multiple initial configurations to the same system of interest. | oc20_dense_mappings.tar.gz |
Validating energy predictions
As mentioned we will be releasing ML relaxed structures along with initial configurations to reduce the computational burden for the challenge. Below are the OC20-Dense-OOD-val ML relaxed structures for Gemnet-OC, SCN, and eSCN in LMDB format. You can make predictions on these structures to validate your approach and compute the success rate without running any DFT on your end.Steps to compute success rate:
- Run energy predictions on ML relaxed structures using one of the model LMDBs below.
- Write predictions to an npz file. This is done automatically if using the OCP codebase. If using a different codebase, a sample npz can be obtained here to help structure your prediction file (Note - this sample was obtained from GemNet-OC-2M, you may have more or less ids depending on the model used).
- Run challenge_eval.py script with npz file to compute success. Details on how to run the script can be found here.
| Name | Description | Download Link |
|---|---|---|
| GemNet-OC-S2EF-2M | LMDB containing 11508 GemNet-OC-S2EF-2M relaxed structures for the OC20-Dense OOD 200 split. All invalid structures have been removed. | gemnet_oc_2M_oc20dense_val_ood.tar.gz |
| SCN-S2EF-2M | LMDB containing 11630 SCN-S2EF-2M relaxed structures for the OC20-Dense OOD 200 split. All invalid structures have been removed. | scn_2M_oc20dense_val_ood.tar.gz |
| eSCN-S2EF-2M | LMDB containing 11755 eSCN-S2EF-2M relaxed structures for the OC20-Dense OOD 200 split. All invalid structures have been removed. | escn_2M_oc20dense_val_ood.tar.gz |
Challenge test data
Participants are to predict the adsorption energy for a total of 200 unique adsorbate+surface combinations. We enumerate ~100 initial configurations for each unique combination resulting in around ~20,000 initial structures. If you wish to generate your own initial structures or have an approach that does not rely on a brute force enumeration of initial configurations, we provide the unique adsorbate and surface atoms objects directly.
Submission Guidelines
To participate in the Open Catalyst Challenge, create a team on EvalAI and upload submissions to the "Predicting adsorption energy given an adsorbate and a catalyst surface -- Test-challenge-2023" phase. Details on how to make a submission can be found here.
Submissions must be an `.npz` numpy binary file in the following format:
{
"ids": array(['64_2752_17', '24_467_11', ...]),
"energy": array([0.13540655, -3.29241061, -3.28643918, ...])
"pos": array([array([[3.18199658e+00, 1.38080716e-01, 2.79435139e+01], [...], [...]]), ...])
}
where `ids`, `energy`, and `pos` are arrays of size 200.
>>> data["ids"].dtype, data["ids"].shape
(dtype('<U12'), (200,))
>>> data["energy"].dtype, data["energy"].shape
(dtype('float64'), (200,))
>>> data["pos"].dtype, data["pos"].shape
(dtype('object'), (200,))
`ids` are the “system_id” key from the challenge_test_2023_lmdb_metadata dictionary, `energy` are the predicted adsorption energies, and `pos` are the predicted relaxed positions.
A few important notes on submissions:
- Participants must provide predictions for each of the 200 unique `system_ids`. We ask that you include predictions even if all configurations for given system_id are determined not to be valid based on the constraints above.
- The validity checks (desorption, dissociation, surface mismatch) assume that the ordering of atoms is consistent with the officially released structures above. So, please ensure that ordering has not been changed.
- For evaluation consistency, we ask that energy predictions are written as float32 or float64.
Our baseline EquiformerV2-2M submission file is provided here as a sample. Please use this for verification / debugging purposes.
Support
Weekly office hours will be held starting July 20, 2023 and run through the duration of the competition. Members of the team will be present to clarify details of the challenge, answer questions, and provide general guidance for those interested in participating.
- Wednesdays, 9:00 - 10:00 AM PT
- Thursdays, 4:00 - 5:00 PM (16:00 - 17:00) PT
If you have any additional questions or concerns regarding the challenge reach out to us via email [1, 2].
The OC20 dataset paper has more details on how the OC20 dataset was created, the various tasks and evaluation metrics, and performance of baseline ML algorithms. The paper is accompanied by our constantly-evolving OCP codebase that provides implementations of several state-of-the-art graph neural network algorithms. The AdsorbML paper has more details on the OC20-Dense dataset, evaluation metrics, and the global minima energy task in general. The paper is accompanied by our adsorbml codebase that provides implementations on identifying physical constraints and evaluation code for both validated and unvalidated success rates. Please consider citing the following if you use it in your work:
@article{lan2022adsorbml,
title={AdsorbML: Accelerating Adsorption Energy Calculations with Machine Learning},
author={Lan*, Janice and Palizhati*, Aini and Shuaibi*, Muhammed and Wood*, Brandon M and Wander, Brook and Das, Abhishek and Uyttendaele, Matt and Zitnick, C Lawrence and Ulissi, Zachary W},
journal={arXiv preprint arXiv:2211.16486},
year={2022}
}
@article{ocp_dataset,
author = {Chanussot*, Lowik and Das*, Abhishek and Goyal*, Siddharth and Lavril*, Thibaut and Shuaibi*, Muhammed and Riviere, Morgane and Tran, Kevin and Heras-Domingo, Javier and Ho, Caleb and Hu, Weihua and Palizhati, Aini and Sriram, Anuroop and Wood, Brandon and Yoon, Junwoong and Parikh, Devi and Zitnick, C. Lawrence and Ulissi, Zachary},
title = {Open Catalyst 2020 (OC20) Dataset and Community Challenges},
journal = {ACS Catalysis},
year = {2021},
doi = {10.1021/acscatal.0c04525},
}
NO PURCHASE NECESSARY TO ENTER/WIN. A PURCHASE WILL NOT INCREASE YOUR CHANCES OF WINNING. Submission Period begins September 20, 2023 at 12:00:00 am UTC and ends October 13, 2023 at 11:59:59 pm UTC. Open to legal residents of the Territory, 18+ & age of majority. "Territory" means any area, country, state, territory, or province where United States or local laws do not prohibit participating or receiving a prize in the Challenge and excludes Russia and any country or jurisdiction that is the target of U.S., EU, United Nations, or UK comprehensive trade sanctions (e.g., Crimea, Donetsk, and Luhansk regions of Ukraine, Cuba, North Korea, Iran, and Syria, as such list may be amended). Void outside the Territory and where prohibited by law. Participation subject to Official Rules. See Official Rules for entry requirements, judging criteria and full details. Winners are invited to attend & present at the Open Catalyst Challenge workshop at NeurIPS in December 2023. Winners are responsible for all costs to attend workshop/conference, including conference registration fee. Sponsors: Meta Platforms, Inc., 1 Hacker Way, Menlo Park, CA 94025 USA and Carnegie Melon University., 5000 Forbes Ave, Pittsburgh, PA 15213.
Overview
The Open Catalyst Challenge 2022 invites participants to help in addressing the pressing challenges faced by the world due to energy scarcity and climate change. In this area, a critical problem is the discovery of new catalysts for driving efficient and carbon neutral means for energy storage and generation. A common approach in discovering high performance catalysts is using molecular simulations, where simpler surrogate descriptors are generated to correlate with experimental measurements of catalyst activity and selectivity. The task for this year's challenge is to design new machine learning models to predict the outcome of catalyst simulations used to understand activity.
Specifically, each simulation models the interaction of a catalyst surface with adsorbates that are commonly seen in electrochemical reactions. The simulations correspond to local relaxations of the atomic positions to identify local minima. ML models are trained to predict the energies of the adsorbate-catalyst system at the local minima, known as the “relaxed state”, starting from a provided initial state. By predicting these interactions accurately, the catalyst's impact on the overall rate of a chemical reaction may be estimated; a key factor in filtering potential electrocatalysis materials and addressing the world's energy needs.
This is the 2nd edition of the Open Catalyst Challenge. The 1st edition was held last year, and the results announced at NeurIPS 2021. More details about last year's challenge can be found here.
This year's challenge focuses on the same task -- Initial Structure to Relaxed Energy (IS2RE) -- as last year (details here). The primary differences are: 1) instead of two tracks, we will have a single track where using the IS2RE data and/or the Structure-to-Energy-Forces (S2EF) 2M training data is allowed (details here). 2) A new test-challenge split will be released in September specifically for this year's challenge.
Dates
Jun 15, 2022: Competition announced
Sep 21, 2022: Challenge test dataset release
Oct 07, 2022: Submission deadline
Oct 21, 2022: Winners notified
Nov 10, 2022: Presenters' slides and videos due
Dec 08, 2022: Winners' announcement and presentations @ NeurIPS, 2022
NeurIPS session
The Open Catalyst Challenge session at NeurIPS 2022 was held on Thursday, December 08, 2022.
The entire session was recorded and is viewable here.
Schedule
| 15:00 - 15:05 CST | Buffer for attendees to join Zoom [room link] | - |
| 15:05 - 15:20 CST | Challenge overview, results and analysis [video] |
Abhishek Das (Open Catalyst Project team)

|
| 15:20 - 15:50 CST | Invited talk + Q&A [video] |

|
| 15:50 - 16:10 CST | Runner-up talk + Q&A [video] |
Yi-Lun Liao, Tess Smidt
(Atomic Architects, MIT) 

|
| 16:10 - 16:30 CST | Winner talk + Q&A [video] |
Jiaqi Han, Tian Bian, Geyan Ye, Kaili Ma, Yuduo Zhi, Kangfei Zhao, Tingyang Xu, Wenbing Huang, Yu Rong
(Tencent AI Lab, Tsinghua University, Renmin University of China, The Chinese University of Hong Kong) 








|
| 16:30 - 17:00 CST | Invited talk + Q&A [video] |

|
| 17:00 - 17:30 CST | Discussion [video] | Attendees, participants, organizers |
Challenge results
The Open Catalyst Challenge received 25 submissions in total from 6 teams. All submissions were evaluated on the test-challenge-2022 dataset split consisting of the following 4 subsplits:
- test-like: similar to OC20 test and used to pick winners
- rotated: used to evaluate rotational invariance
- anomalous: structures with desorptions and dissociations
- dense: dense sampling of adsorbate placements for evaluating recall of lowest energy site
| Energy MAE (eV) | |||||
|---|---|---|---|---|---|
| Rank | Team | Test-like | Rotated | Anomalous | Dense |
| 1 |
TTRC (previously "Tencent AI Lab") Jiaqi Han1, Tian Bian2, Geyan Ye3, Kaili Ma2, Yuduo Zhi3, Kangfei Zhao3, Tingyang Xu3, Wenbing Huang4, Yu Rong3 1=Tsinghua University, 2=The Chinese University of Hong Kong, 3=Tencent AI Lab, 4=Renmin University of China |
0.3960 | 0.3964 | 0.8904 | 0.3630 |
| 2 |
Atomic Architects MIT Yi-Lun Liao, Tess Smidt Massachusetts Institute of Technology |
0.4266 | 0.4235 | 0.9228 | 0.3838 |
| 3 |
XJTUNRTeam |
0.5255 | 0.5176 | 1.1577 | 0.4718 |
| 3 |
AutoGraph Xu Wang, Huan Zhao 4Paradigm |
0.5263 | 0.5176 | 1.0789 | 0.4933 |
| 5 |
Shanghai Jiao Tong University Wei Yang1, Frank Ji1, Yulian He2, Cheng Hua3, Guanjie Zheng3, Zhanyu Liu3, Feixiang Tian2, Tianhua Li3, Junlin He3 1=Yalotein Biotech, 2=University of Michigan - Shanghai Jiao Tong University Joint Institute, 3=Shanghai Jiao Tong University |
0.6529 | 0.6603 | 1.2163 | 0.5875 |
| 6 |
personal test Bangjian Zhou1, Ji Wei Yoon1, Zhuoyi Lin1, J Senthilnath1, Chaitanya K. Joshi2 1=Agency for Science, Technology and Research, Singapore, 2=University of Cambridge |
1.0709 | 1.0671 | 1.5259 | 0.6058 |
IS2RE Task
The challenge will consist of one primary task -- Initial Structure to Relaxed Energy (IS2RE) [1]. Here the input consists of the atomic positions for an initial structure, and the goal is to predict the energy of the structure's relaxed state.
Relaxed energies are a critical indicator in determining the reaction rate resulting from the use of a catalyst. By placing an adsorbate in multiple locations above a catalyst's surface and relaxing the structure, the binding site between the adsorbate and catalyst with the lowest relaxed energy can be determined. This lowest energy binding site is likely to be the one realized in practice under experimental conditions. The relaxed energy of the lowest energy binding site is also highly correlated with the reaction rates or selectivity of the chemical reaction. If successful, these techniques could be used to screen millions or even billions of potential catalyst materials for the chemical reactions involved in renewable energy storage and solar fuel generation.
Traditionally, relaxed energies are found by first performing structure relaxations through an iterative local optimization process that estimates the gradients (atomic forces) using Density Functional Theory (DFT), which are in turn used to update atom positions until convergence. This very computationally expensive process typically requires hundreds of DFT calculations to converge (hours or days of compute per relaxation) and forms the basis of most computational catalysis efforts.
One approach to the IS2RE task is relaxation-based, i.e. using ML to approximate DFT relaxations. These models iteratively estimate atomic forces and update atomic positions until a relaxed state is reached and finally predict the energy of that state. Evaluation of the IS2RE task on models built for approximating DFT relaxations will help determine whether this approach is sufficiently accurate and fast for practical applications. These models have the additional benefit of predicting the relaxed structure and accelerating future DFT calculations.
Alternatively, it may be possible to develop direct approaches that predict the relaxed energy directly, without estimating intermediate relaxation states, as many of the changes during a relaxation (say due to particular initial guess strategies) are systematic. These direct IS2RE approaches may lead to even greater improvements in computational efficiency.
As such, we place no restrictions on the possible ML approaches to solve this task and used to participate in this challenge. We encourage submissions that are significantly more computationally efficient than DFT. For example, a standard relaxation using DFT takes 8-10 hours, while ML approaches are desired that can bring this down to < 10 seconds per relaxation or < 1 second per direct prediction, at least a 1000x improvement!
To ensure consistent and fair evaluation, we use a public evaluation server hosted on EvalAI.
Dataset
The challenge will be conducted on the Open Catalyst Dataset (OC20). OC20 training and validation data are available here. A new test-challenge-2022 split has been released here specifically for this challenge. This is to ensure there is no overfitting on the test data through repeated submissions.
OC20 contains approximately ~1.2M DFT relaxations. Due to its significant scale, the dataset required over ~200M hours of compute to generate. Computation was performed on servers Meta has committed to be 100% supported by renewable energy since 2020. Each relaxation contains a series of structures as the atoms move from an initial structure to a relaxed structure obtained through a standard local minimizer built in the computational chemistry code. Structures contain the atoms corresponding to the adsorbate and catalyst. The initial structures are heuristically determined and the relaxed structures correspond to a state in which the atoms are at a local energy minima. Since each step in the relaxation may be used for training / evaluation, the total number of simulation points is over ~264M! The largest training split has ~134M simulation points. However, for this challenge, only the 2M dataset may be used for training. For each structure, DFT computed system energies, per-atom forces and per-atom positions are available as annotations.
The OC20 validation and test splits have several subsplits to help evaluate a model's performance on interpolative and extrapolative tasks. A model's interpolative ability is evaluated on samples from the same distribution as the training dataset (In Domain). Extrapolation is evaluated on two dimensions -- new adsorbates and new catalyst compositions. Subsplits are created by considering all combinations of potential extrapolations -- Out-of-Domain Adsorbate (OOD Adsorbate), OOD Catalyst, and OOD Both (both unseen adsorbate and unseen catalyst compositions).
Summary of all evaluation splits
| Split | Size | Max submissions | Metrics | Results | Leaderboard |
|---|---|---|---|---|---|
| val | ~100k | - | Energy MAE, EwT | On EvalAI | - |
| test | ~100k | 10 | Energy MAE, EwT | On EvalAI | On opencatalystproject.org all year round |
| test-challenge-2022 | ~100k | 10 | Energy MAE, EwT | Will be announced at NeurIPS '22 | Will be announced at NeurIPS '22 |
Evaluation
All submissions to the Open Catalyst Challenge will be made to the EvalAI server and evaluated on the following metrics:
- Energy MAE: mean absolute error between the predicted relaxed energy and the DFT-computed ground-truth relaxed energy.
- Energy within Threshold (EwT): the percentage of predicted relaxed energies within 0.02 eV of the DFT-computed ground-truth relaxed energy.
Challenge winners will be decided based on the Energy MAE metric.
The challenge will have a single track, wherein participants are allowed to train on the IS2RE dataset (size 460k) and/or the S2EF 2M dataset. We are expanding the scope from last year to include and encourage training on intermediate trajectory data from S2EF (in addition to IS2RE direct data) because we have seen that to consistently improve performance. We acknowledge that resource availability may become a bottleneck for some participants and hence will not be accepting entries that train on S2EF splits larger than 2M.
The table below summarizes training compute costs and accuracies of various direct and relaxation-based IS2RE baseline approaches. Training a relaxation-based GemNet-OC S2EF-2M model is about twice as expensive as training a direct Graphormer model, but improves energy MAE by ~20%! Training on S2EF-All (in red) improves accuracies further but gets prohibitively expensive, and hence training on S2EF-All is not allowed for challenge entrants. Our non-challenge test evaluation server and leaderboards are open all year round for these larger trained models. The North star of this challenge will be to marry the accuracy of relaxation-based models with the efficiency of direct models.
| Model | IS2RE approach | Training dataset | Training GPU hours | Test Energy MAE |
|---|---|---|---|---|
|
GemNet-dT (paper, code) |
Direct | IS2RE | 75 | 0.634 |
|
PaiNN (paper, code) |
Direct | IS2RE | 518 | 0.573 |
|
Graphormer Open Catalyst Challenge 2021 winner (paper, code) |
Direct | IS2RE | 568 | 0.538 |
|
GemNet-dT (paper, code) |
Relaxation | S2EF-2M | 863 | 0.438 |
|
GemNet-OC (paper, code) |
Relaxation | S2EF-2M | 1183 | 0.407 |
|
PaiNN (paper, code) |
Relaxation | S2EF-All | 1600 | 0.471 |
|
GemNet-dT (paper, code) |
Relaxation | S2EF-All | 11820 | 0.400 |
|
GemNet-OC (paper, code) |
Relaxation | S2EF-All | 8067 | 0.355 |
Participants will be prompted while making submissions to EvalAI to confirm that they didn't use any training data outside of the IS2RE and the S2EF-2M datasets. Data augmentation and pretraining is permitted as long as it comes only from the IS2RE or S2EF-2M datasets. Using DFT is not allowed.
We will be inviting the winner and runner-up teams, and optionally teams with interesting entries (e.g. best direct approach) for oral presentations at NeurIPS 2022.
Submission Guidelines
To participate in the Open Catalyst Challenge, create a team on EvalAI and upload submissions to the "Predicting relaxed state energy from initial structure (IS2RE) -- Test-challenge-2022" phase:
Submissions must be an `.npz` numpy binary file in the following format:
{
"challenge_ids": array(['0', '1', ...]),
"challenge_energy": array([-3.63920, -1.08237, 12.92103, ...,])
}
where both `challenge_ids` and `challenge_energy` are arrays of size 100010.
>>> data["challenge_ids"].dtype, data["challenge_ids"].shape
(dtype('<U6'), (100010,))
>>> data["challenge_energy"].dtype, data["challenge_energy"].shape
(dtype('float64'), (100010,))
A dummy submission file is available here.
Please use this for verification / debugging purposes.
We also provide helper code and docs for training models and preparing
EvalAI submission files for the IS2RE task here.
The OC20 dataset paper has more details on how the OC20 dataset was created, the various tasks and evaluation metrics, and performance of baseline ML algorithms. The paper is accompanied by our constantly-evolving OCP codebase that provides implementations of several state-of-the-art graph neural network algorithms. Please consider citing the following if you use it in your work:
@article{ocp_dataset,
author = {Chanussot*, Lowik and Das*, Abhishek and Goyal*, Siddharth and Lavril*, Thibaut and Shuaibi*, Muhammed and Riviere, Morgane and Tran, Kevin and Heras-Domingo, Javier and Ho, Caleb and Hu, Weihua and Palizhati, Aini and Sriram, Anuroop and Wood, Brandon and Yoon, Junwoong and Parikh, Devi and Zitnick, C. Lawrence and Ulissi, Zachary},
title = {Open Catalyst 2020 (OC20) Dataset and Community Challenges},
journal = {ACS Catalysis},
year = {2021},
doi = {10.1021/acscatal.0c04525},
}
NO PURCHASE NECESSARY TO ENTER/WIN. A PURCHASE WILL NOT INCREASE YOUR CHANCES OF WINNING. Submission Period begins September 21, 2022 at 12:00:00 am UTC and ends October 7, 2022 at 11:59:59 pm UTC. Open to legal residents of the Territory, 18+ & age of majority. "Territory" means any area, country, state, territory, or province where United States or local laws do not prohibit participating or receiving a prize in the Challenge and excludes any country or jurisdiction that is the target of U.S., EU, United Nations, or UK comprehensive trade sanctions (e.g., Crimea, Donetsk, and Luhansk regions of Ukraine, Cuba, North Korea, Iran, and Syria, as such list may be amended). Void outside the Territory and where prohibited by law. Participation subject to Official Rules. See Official Rules for entry requirements, judging criteria and full details. Winners are invited to attend & present at the Open Catalyst Challenge workshop at NeurIPS in December 2022. Winners are responsible for all costs to attend workshop/conference, including conference registration fee. Sponsors: Meta Platforms, Inc., 1 Hacker Way, Menlo Park, CA 94025 USA and Carnegie Melon University., 5000 Forbes Ave, Pittsburgh, PA 15213.
Overview
The Open Catalyst Challenge invites participants to help in addressing the pressing challenges faced by the world due to energy scarcity and climate change. In this area, a critical problem is the discovery of new catalysts for driving efficient and carbon neutral means for energy storage and generation. A common approach in discovering high performance catalysts is using molecular simulations, where simpler surrogate descriptors are generated to correlate with experimental measurements of catalyst activity and selectivity. The task for this year’s challenge is to design new machine learning models to predict the outcome of catalyst simulations used to understand activity.
Specifically, each simulation models the interaction of a catalyst surface with adsorbates that are commonly seen in electrochemical reactions. The simulations correspond to local relaxations of the atomic positions to identify local minima. ML models are trained to predict the energies of the adsorbate-catalyst system at the local minima, known as the “relaxed state”, starting from a provided initial state. By predicting these interactions accurately, the catalyst's impact on the overall rate of a chemical reaction may be estimated; a key factor in filtering potential electrocatalysis materials and addressing the world's energy needs.
Dates
Jun 17, 2021: Competition announced
Sep 20, 2021: Challenge test dataset release
Oct 06, 2021: Submission deadline
Oct 20, 2021: Winners notified
Nov 10, 2021: Presenters' slides and videos due
Dec 07, 2021: Winners' announcement and presentations @ NeurIPS, 2021
NeurIPS session
The Open Catalyst Challenge session at NeurIPS 2021 is scheduled for Tuesday, December 07, 2021, 18:05 GMT (or 10:05 AM PST) onwards. Note that it is mandatory to register for NeurIPS to attend the session.
The session starts with a broadcast of the challenge overview and results at 18:05 GMT on the NeurIPS website here, followed by a breakout session on Zoom.
Schedule
| 18:05 - 18:25 GMT | Challenge overview, results and analysis [video] | Abhishek Das, Muhammed Shuaibi, Aini Palizhati |
| 18:25 - 18:30 GMT | Buffer for attendees to join Zoom [room link] | - |
| 18:30 - 18:50 GMT | Runner-up talk + Q&A [video] | Innopolis AI |
| 18:50 - 19:10 GMT | Winner talk + Q&A [video] | Microsoft Research Asia (previously "MachineLearning") |
| 19:10 - 19:30 GMT | Discussion [video] | Attendees, participants, organizers |
Challenge results
The Open Catalyst Challenge received 30 submissions in total from 7 teams. All submissions were to the IS2RE-only track, and were evaluated on the test-challenge dataset split consisting of the following 4 subsplits:
- test-like: similar to OC20 test and used to pick winners
- rotated: used to evaluate rotational invariance
- anomalous: structures with desorptions and dissociations
- dense: dense sampling of adsorbate placements for evaluating recall of lowest energy site
| Energy MAE (eV) | |||||
|---|---|---|---|---|---|
| Rank | Team | Test-like | Rotated | Anomalous | Dense |
| 1 |
Microsoft Research Asia (previously "MachineLearning") Guolin Ke1, Chengxuan Ying2, Shuxin Zheng1, Di He1, Jiacheng You3, Yihan He4 1=Microsoft Research Asia, 2=Dalian University of Technology, 3=Tsinghua University, 4=Carnegie Mellon University |
0.5474 | 0.5467 | 1.0312 | 0.6353 |
| 2 |
Innopolis AI Rostislav Grigoriev, Ruslan Lukin, Adel Yarullin, Max Faleev Innopolis University, Russia |
0.6180 | 0.6170 | 1.1859 | 0.6839 |
| 3 |
Up and Atom Adam Maximilian Wilson, Sam Walton Norwood, Peter Bjørn Jørgensen Technical University of Denmark |
0.6694 | 0.6707 | 1.1402 | 0.7398 |
| 3 |
DIVE @ TAMU Limei Wang, Yuchao Lin, Xiner Li, Jingtun Zhang, Yi Liu, Shurui Gui, Keqiang Yan, Shuiwang Ji Texas A&M University |
0.6710 | 0.6712 | 1.1810 | 0.7398 |
| 5 |
RedSeaSeed Hao Yu King Abdullah University of Science and Technology |
0.6830 | 0.6811 | 1.1876 | 0.7435 |
| 6 |
air Alexey Korovin, Roman Eremin, Innokentiy Humonen, Artem Vasilyev, Vladimir Lazarev, Semeon Budennyy Artificial Intelligence Research Institute, Moscow |
0.6973 | 0.6999 | 1.3089 | 0.7594 |
| 7 |
EnergyNet Mayank Baranwal1, Nawaf Alampara2, Ravi Bhadauria3 1=Tata Consultancy Services Research and Innovation, Mumbai, Indian Institute of Technology, Bombay, 2=QpiVolta Technologies Pvt. Ltd., Bengaluru, India, 3=Etsy, New York, USA |
0.7351 | 0.8842 | 1.3399 | 0.8033 |
IS2RE Task
The challenge will consist of one primary task -- Initial Structure to Relaxed Energy (IS2RE) [1]. Here the input consists of the atomic positions for an initial structure, and the goal is to predict the energy of the structure’s relaxed state.
Relaxed energies are a critical indicator in determining the reaction rate resulting from the use of a catalyst. By placing an adsorbate in multiple locations above a catalyst's surface and relaxing the structure, the binding site between the adsorbate and catalyst with the lowest relaxed energy can be determined. This lowest energy binding site is likely to be the one realized in practice under experimental conditions. The relaxed energy of the lowest energy binding site is also highly correlated with the reaction rates or selectivity of the chemical reaction. If successful, these techniques could be used to screen millions or even billions of potential catalyst materials for the chemical reactions involved in renewable energy storage and solar fuel generation.
Traditionally, relaxed energies are found by first performing structure relaxations through an iterative local optimization process that estimates the gradients (atomic forces) using Density Functional Theory (DFT), which are in turn used to update atom positions until convergence. This very computationally expensive process typically requires hundreds of DFT calculations to converge (hours or days of compute per relaxation) and forms the basis of most computational catalysis efforts.
One approach to the IS2RE task is using ML to approximate DFT relaxations i.e. iteratively estimate atomic forces and update atomic positions until a relaxed state is reached and finally predict the energy of that state. Evaluation of the IS2RE task on models built for approximating DFT relaxations will help determine whether this approach is sufficiently accurate and fast for practical applications. These models have the additional benefit of predicting the relaxed structure and accelerating future DFT calculations. Alternatively, it may be possible to predict the relaxed energy directly, without estimating intermediate relaxation states, as many of the changes during a relaxation (say due to particular initial guess strategies) are systematic. These direct IS2RE approaches may lead to even greater improvements in computational efficiency. As such, we place no restrictions on the possible ML approaches to solve this task and used to participate in this challenge. We encourage submissions that are significantly more computationally efficient than DFT. For example, a standard relaxation using DFT takes 8-10 hours, while ML approaches are desired that can bring this down to < 10 seconds per relaxation or < 1 second per direct prediction, at least a 1000x improvement!
To ensure consistent and fair evaluation, we use a public evaluation server hosted on EvalAI.
Dataset
The challenge will be conducted on the Open Catalyst Dataset (OC20). OC20 training and validation data are available here. A new test-challenge split has been released here specifically for this challenge. This is to ensure there is no overfitting on the test data through repeated submissions.
OC20 contains approximately ~1.2M DFT relaxations. Due to its significant scale, the dataset required over ~70M hours of compute to generate. Computation was performed on servers Meta has committed to be 100% supported by renewable energy since 2020. Each relaxation contains a series of structures as the atoms move from an initial structure to a relaxed structure obtained through a standard local minimizer built in the computational chemistry code. Structures contain the atoms corresponding to the adsorbate and catalyst. The initial structures are heuristically determined and the relaxed structures correspond to a state in which the atoms are at a local energy minima. Since each step in the relaxation may be used for training / evaluation, the total number of simulation points is over ~264M! The largest training split has ~134M simulation points. For each structure, DFT computed system energies, per-atom forces and per-atom positions are available as annotations.
The OC20 validation and test splits have several subsplits to help evaluate a model's performance on interpolative and extrapolative tasks. A model's interpolative ability is evaluated on samples from the same distribution as the training dataset (In Domain). Extrapolation is evaluated on two dimensions -- new adsorbates and new catalyst compositions. Subsplits are created by considering all combinations of potential extrapolations -- Out-of-Domain Adsorbate (OOD Adsorbate), OOD Catalyst, and OOD Both (both unseen adsorbate and unseen catalyst compositions).
Summary of all evaluation splits
| Split | Size | Max submissions | Metrics | Results | Leaderboard |
|---|---|---|---|---|---|
| val | ~100k | - | Energy MAE, EwT | On EvalAI | - |
| test | ~100k | 10 | Energy MAE, EwT | On EvalAI | On opencatalystproject.org all year round |
| test-challenge | 120k | 10 | Energy MAE, EwT | Announced at NeurIPS '21 | Announced at NeurIPS '21 |
Evaluation
All submissions to the Open Catalyst Challenge will be made to the EvalAI server and evaluated on the following metrics:
- Energy MAE: mean absolute error between the predicted relaxed energy and the DFT-computed ground-truth relaxed energy.
- Energy within Threshold (EwT): the percentage of predicted relaxed energies within 0.02 eV of the DFT-computed ground-truth relaxed energy.
Challenge winners will be decided based on the Energy MAE metric.
We acknowledge that resource availability may become a bottleneck for some participants given the large size of the OC20 trajectory data (~134M training points). Thus, we will be recognizing 2 winners for the challenge based on:
- The best overall performance with no constraints on data used
- The best performance using ONLY the IS2RE dataset (size 460,328)
Submission Guidelines
To participate in the Open Catalyst Challenge, create a team on EvalAI and upload submissions to the "Predicting relaxed state energy from initial structure (IS2RE) -- Test-challenge" phase:
Submissions must be an `.npz` numpy binary file in the following format:
{
"challenge_ids": array(['0', '1', ...]),
"challenge_energy": array([-3.63920, -1.08237, 12.92103, ...,])
}
where both `challenge_ids` and `challenge_energy` are arrays of size 120000.
>>> data["challenge_ids"].dtype, data["challenge_ids"].shape
(dtype('<U6'), (120000,))
>>> data["challenge_energy"].dtype, data["challenge_energy"].shape
(dtype('float64'), (120000,))
A dummy submission file is available here.
Please use this for verification / debugging purposes.
We also provide helper code and docs for training models and preparing
EvalAI submission files for the IS2RE task here.
The OC20 dataset paper has more details on how the OC20 dataset was created, the various tasks and evaluation metrics, and performance of baseline ML algorithms. The paper is accompanied by our constantly-evolving OCP codebase that provides implementations of several state-of-the-art graph neural network algorithms. Consider citing the following if you use it in your work:
@article{ocp_dataset,
author = {Chanussot*, Lowik and Das*, Abhishek and Goyal*, Siddharth and Lavril*, Thibaut and Shuaibi*, Muhammed and Riviere, Morgane and Tran, Kevin and Heras-Domingo, Javier and Ho, Caleb and Hu, Weihua and Palizhati, Aini and Sriram, Anuroop and Wood, Brandon and Yoon, Junwoong and Parikh, Devi and Zitnick, C. Lawrence and Ulissi, Zachary},
title = {Open Catalyst 2020 (OC20) Dataset and Community Challenges},
journal = {ACS Catalysis},
year = {2021},
doi = {10.1021/acscatal.0c04525},
}
NO PURCHASE NECESSARY TO ENTER/WIN. A PURCHASE WILL NOT INCREASE YOUR CHANCES OF WINNING. Submission Period begins September 20, 2021 at 12:00:00 am UTC and ends October 6, 2021 at 11:59:59 pm UTC. Open to legal residents of the Territory, 18+ & age of majority. "Territory" means any country, state, or province where the laws of the US or local law do not prohibit participating or receiving a prize in the Challenge and excludes Cuba, Crimea, North Korea, Iran, Syria, Venezuela and any other jurisdiction or area designated by the United States Treasury's Office of Foreign Assets Control. Void outside the Territory and where prohibited by law. Participation subject to Official Rules. See Official Rules for entry requirements, judging criteria and full details. Winners are invited to attend & present at the virtual Open Catalyst Challenge session at NeurIPS on December 13 or 14, 2021. Winners are responsible for all costs to attend workshop/conference, including conference registration fee. Sponsor: Facebook, Inc., 1 Hacker Way, Menlo Park, CA 94025 USA.